reacción de inversión de quiralidad puede cambiar completamente las estrategias de síntesis total | Búsqueda
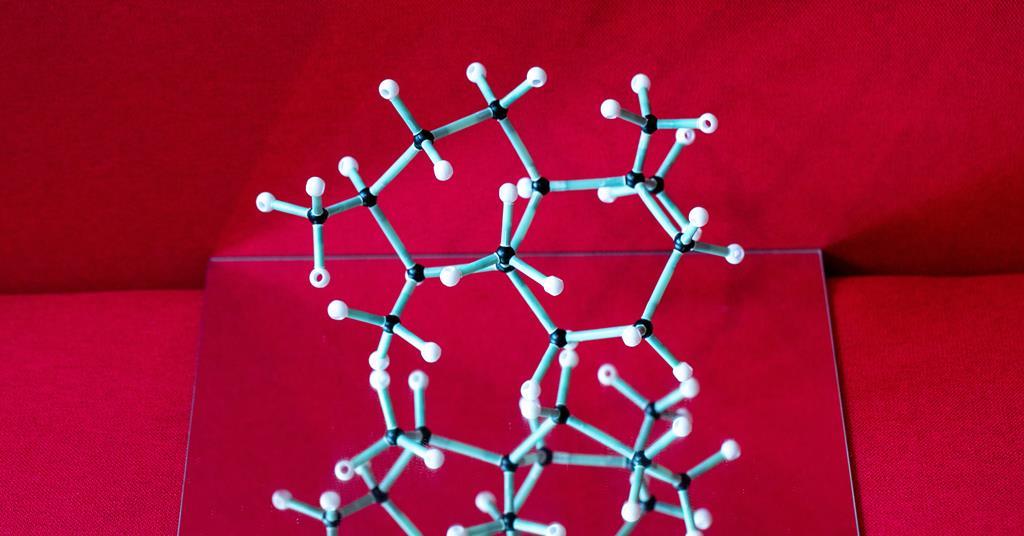
Los investigadores han desarrollado una reacción que convierte moléculas quirales en el estereoisómero opuesto. Funciona sobre estereocentros no activados que hasta ahora eran difíciles de alcanzar. El método podría permitir a los investigadores acceder a nuevas configuraciones moleculares y podría cambiar la forma en que los químicos planifican sus síntesis totales.
Cuando los químicos deciden construir moléculas quirales complejas, tienden a utilizar secuencias de reacción diseñadas en torno a la construcción de centros quirales. Este es a menudo el aspecto más desafiante de la complejidad del edificio durante un proyecto de síntesis, señala Alison Wendlandt del Instituto Tecnológico de Massachusetts, Estados Unidos.
Ahora, el equipo de Wendlandt ha encontrado una manera de construir diastereómeros de moléculas complejas similares a fármacos de una manera estratégicamente diferente. “Nuestra herramienta esencialmente nos permite intentar ajustar la estructura tridimensional en un escenario de etapa avanzada”, explica. ‘Y entonces [if] tiene un diastereómero, ahora puede acceder a un diastereómero completamente diferente sin tener que volver a sintetizar la molécula con la estereoforma correcta que deseaba.’ Esto significa que al planificar una síntesis, los químicos ahora pueden desacoplar los pasos que forman enlaces o generan complejidad de los pasos necesarios para lograr el control tridimensional, dice Wendlandt.
La técnica se basa en un fotocatalizador polianiónico de tungsteno y cocatalizadores de disulfuro que promueven reacciones de epimerización, como las que invierten la configuración de un centro quiral. equipo de wendland estereoquímica previamente alterada en azúcaresy ahora ha demostrado que funciona en una gama mucho más amplia de moléculas que contienen estereocentros de carbono terciario no activado.
«Donde creo que esto realmente brillará es en la aplicación de la química médica», dice Wendlandt. “Es muy difícil predecir cuál será el efecto de un cierto estereocentro nuevamente en la interacción de esa pequeña molécula con, digamos, una enzima o proteína, y luego, francamente, solo tenemos que hacer todos los diastereoisómeros de una molécula quiral y mira lo que pasa’.
Usando esta lógica de edición estereoscópica, los químicos de descubrimiento de fármacos podrían simplemente sintetizar el diastereómero más fácil de hacer y luego acceder rápidamente al isómero opuesto usando el fotocatalizador de Wendlandt. ‘Incluso si obtiene un rendimiento del 10% o 20% de un objetivo, eso es suficiente para validar: ¿es este un mejor candidato a fármaco? ¿O es un peor candidato a fármaco? Y eso puede ahorrar mucho tiempo”, dice Wendlandt.
Universidad de Wisconsin–Madison, EE. UU., especialista en fotocatálisis Tehshik Yoon describe los resultados como «notable». Señala que si bien existen numerosas estrategias para epimerizar los estereocentros cercanos a los grupos funcionales polares, «la capacidad de cambiar la estereoquímica en un carbono no funcionalizado en una molécula compleja es extraordinaria».
“Creo que esto puede afectar la química sintética de varias maneras, pero lo que más me emociona es la forma en que descarrila los métodos que producen relaciones estereoquímicas bien establecidas. [for example] la reacción de Diels-Alder, hacia resultados estereoquímicos que de otro modo serían muy difíciles de producir’, añade Yoon. «El hecho de que el método utilice reactivos comunes y bien aceptados solo hace que la innovación sea aún más impresionante y probablemente más fácil de adoptar para el resto del campo».