
Un nuevo software permite a los científicos modelar proteínas que cambian de forma en entornos celulares nativos
× cerrar
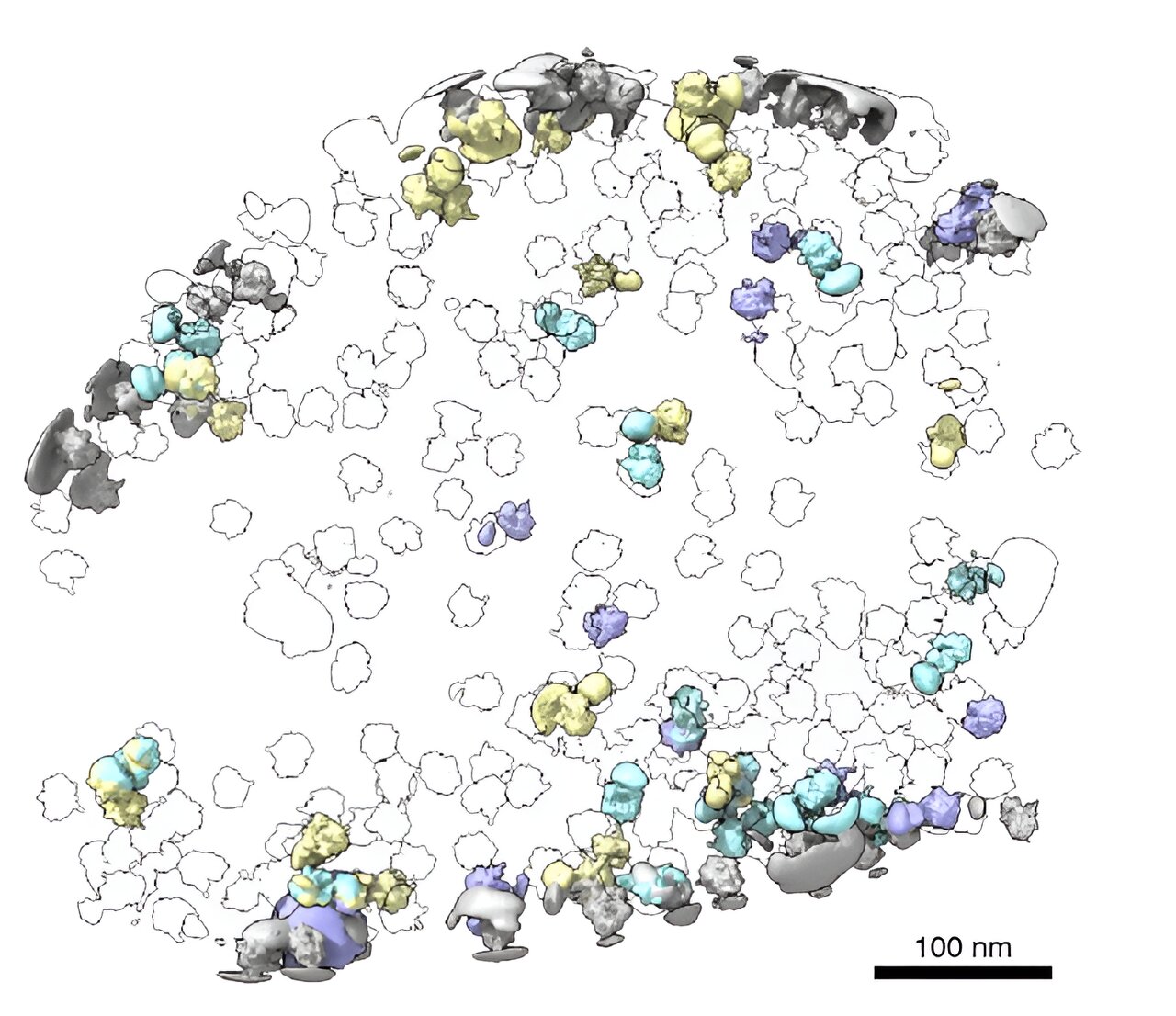
La tomografía electrónica criogénica (crio-ET) es una forma de observar proteínas en su entorno nativo mediante la obtención de imágenes de células congeladas en diferentes ángulos para obtener información estructural en 3D. Esta ilustración muestra cómo una herramienta adicional, desarrollada en el MIT, obtiene imágenes de las estructuras de los ribosomas, revelando las interacciones ribosoma-ribosoma y ribosoma-membrana a partir de datos crio-ET. Crédito: Barrett Powell
Las células dependen de complejas máquinas moleculares formadas por conjuntos de proteínas para realizar funciones esenciales como la producción de energía, la expresión genética y la síntesis de proteínas. Para comprender mejor cómo funcionan estas máquinas, los científicos capturan imágenes de ellas aislando proteínas de las células y utilizando varios métodos para determinar sus estructuras. Sin embargo, este proceso también los elimina del contexto de su entorno nativo, incluidos los compañeros de interacción de proteínas y la ubicación celular.
Recientemente, la tomografía electrónica criogénica (crio-ET) ha surgido como una forma de observar proteínas en su entorno nativo mediante imágenes de células congeladas en diferentes ángulos para obtener información estructural tridimensional. Este enfoque es interesante porque permite a los investigadores observar directamente cómo y dónde se asocian las proteínas entre sí, revelando la vecindad celular de estas interacciones dentro de la célula.
Con la tecnología disponible para obtener imágenes de proteínas en su entorno nativo, el estudiante graduado del MIT Barrett Powell se preguntó si podría ir un paso más allá: ¿Qué pasaría si las máquinas moleculares pudieran observarse en acción? en un papel Publicado 8 de marzo en Los métodos de la naturaleza, Powell describe el método que desarrolló, llamado tomoDRGN, para modelar diferencias estructurales de proteínas en datos crio-ET que surgen de los movimientos de proteínas o de la unión de proteínas a diferentes socios de interacción. Estas variaciones se conocen como heterogeneidad estructural.
Aunque Powell se unió al laboratorio del profesor asociado de biología del MIT Joey Davis como científico experimental, reconoció el impacto potencial de los enfoques computacionales en la comprensión de la heterogeneidad estructural dentro de una célula. Anteriormente, el Laboratorio Davis desarrolló una metodología relacionada llamada crioDRGN para comprender la heterogeneidad estructural en muestras purificadas. Cuando Powell y Davis vieron que la crio-ET ganaba importancia en el campo, Powell asumió el desafío de reinventar esta estructura para que funcione en las células.
Al resolver estructuras con muestras purificadas, se visualiza cada partícula solo una vez. Por otro lado, los datos crio-ET se recopilan tomando imágenes de cada partícula más de 40 veces desde diferentes ángulos. Esto significaba que tomoDRGN necesitaba poder fusionar información de más de 40 imágenes, y ahí es donde el proyecto tuvo un problema: la cantidad de datos provocó una sobrecarga de información.
Para abordar esto, Powell reconstruyó con éxito el modelo crioDRGN para priorizar solo los datos de la más alta calidad. Al observar la misma partícula varias veces, se produce daño por radiación. Por lo tanto, las imágenes adquiridas previamente tienden a ser de mayor calidad porque las partículas están menos dañadas.
«Al excluir algunos de los datos de menor calidad, los resultados fueron en realidad mejores que utilizando todos los datos, y el rendimiento computacional fue sustancialmente más rápido», dice Powell.
Justo cuando Powell comenzaba a probar su modelo, tuvo un golpe de suerte: los autores de un nuevo e innovador estudio que visualizó, por primera vez, ribosomas dentro de las células con una resolución casi atómica compartieron sus datos sin procesar en el Archivo de Imágenes Públicas de Microscopía Eléctrica. (EMPIAR). Este conjunto de datos fue un caso de prueba ejemplar para Powell, a través del cual demostró que tomoDRGN podría descubrir heterogeneidad estructural en los datos crio-ET.
Según Powell, un resultado interesante es lo que tomoDRGN descubrió en torno a un subconjunto de ribosomas en el conjunto de datos EMPIAR. Algunas de las partículas ribosómicas estaban asociadas con una membrana celular bacteriana y participaban en un proceso llamado translocación cotraduccional. Esto ocurre cuando una proteína se sintetiza y transporta simultáneamente a través de una membrana.
Mas informaciones:
Barrett M. Powell et al, Aprendizaje de la heterogeneidad estructural a partir de subtomogramas de crioelectrones con tomoDRGN, Los métodos de la naturaleza (2024). DOI: 10.1038/s41592-024-02210-z
Esta historia se republica por cortesía de MIT News (web.mit.edu/newsoffice/), un sitio web popular que cubre noticias sobre investigación, innovación y enseñanza del MIT.


:quality(70)/cloudfront-eu-central-1.images.arcpublishing.com/thenational/PBF6YAI6HUHEKXLWJBVKHITDZ4.jpg)

