Los subtipos moleculares asociados con la infiltración de células inmunes y la firma genética predicen el pronóstico de los pacientes con osteosarcoma

ML se relacionó con el pronóstico de pacientes con OS
En nuestro estudio, se realizaron el algoritmo contador MCP y el análisis de regresión de Cox univariado y univariado para identificar las células inmunes asociadas con la supervivencia. Los resultados del análisis de regresión de Cox univariante mostraron que ML era una célula inmune relacionada con la supervivencia para los pacientes con OS (Fig. 1A). Además, el análisis de Kaplan-Meier indicó que un nivel bajo de ML se relacionaba con un peor pronóstico para los pacientes con OS (p = 0,033, Fig. 1B).
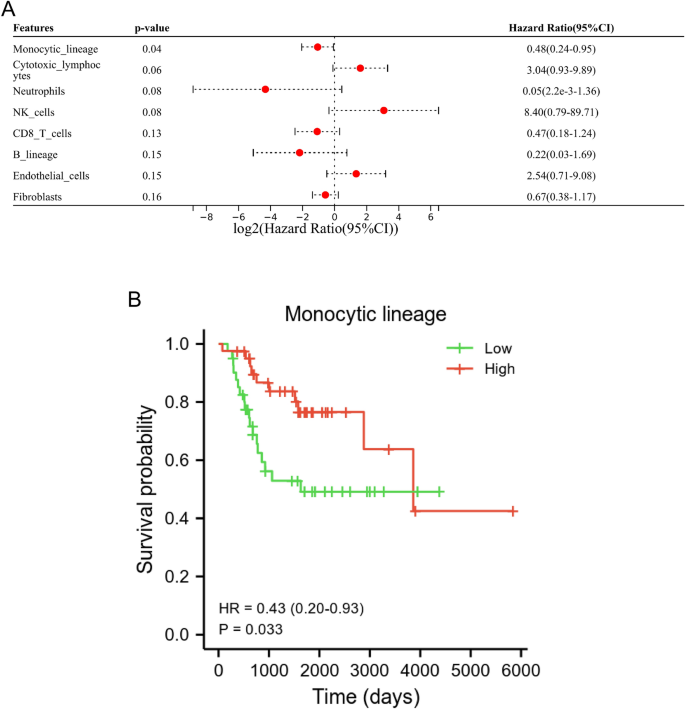
La ML se ha relacionado con un mal pronóstico en pacientes con OS. (A) Análisis univariado de Cox de seis células inmunes y dos células estromales basado en la base de datos TARGET. (B) El análisis de Kaplan-Meier mostró que el nivel de ML se asoció significativamente con el pronóstico de los pacientes.
Análisis de DEG y posibles vías de señalización entre dos subgrupos.
Se identificaron un total de 435 DEG después del análisis de DEG (Fig. 2A, B). En comparación con LML, 101 genes estaban regulados negativamente y 334 genes estaban regulados positivamente en el grupo HML. En el término de procesos biológicos (BP), estos DEG participaron en la regulación de la activación de los linfocitos, la activación de las células T, la regulación positiva de la activación celular, la regulación de la activación de las células T, la adhesión célula-célula de leucocitos, etc. (CC), los DEG estuvieron involucrados en el lado externo de la membrana plasmática, la membrana de los gránulos secretores, los gránulos ricos en girolina-1, la sinapsis inmunológica, el complejo NADPH oxidasa, etc. En términos de funciones moleculares (MF), los DEG se enriquecieron significativamente en la unión a carbohidratos, la unión al receptor de citoquinas, la unión de citocinas, la actividad del receptor de citoquinas, la actividad del receptor de quimiocinas C – C, etc. En términos de KEGG, los DEG se enriquecieron principalmente en el linaje de células hematopoyéticas, la diferenciación de osteoclastos, la interacción citocina-receptor de citocina, la vía de señalización del receptor de células B, etc. (Fig. 3).
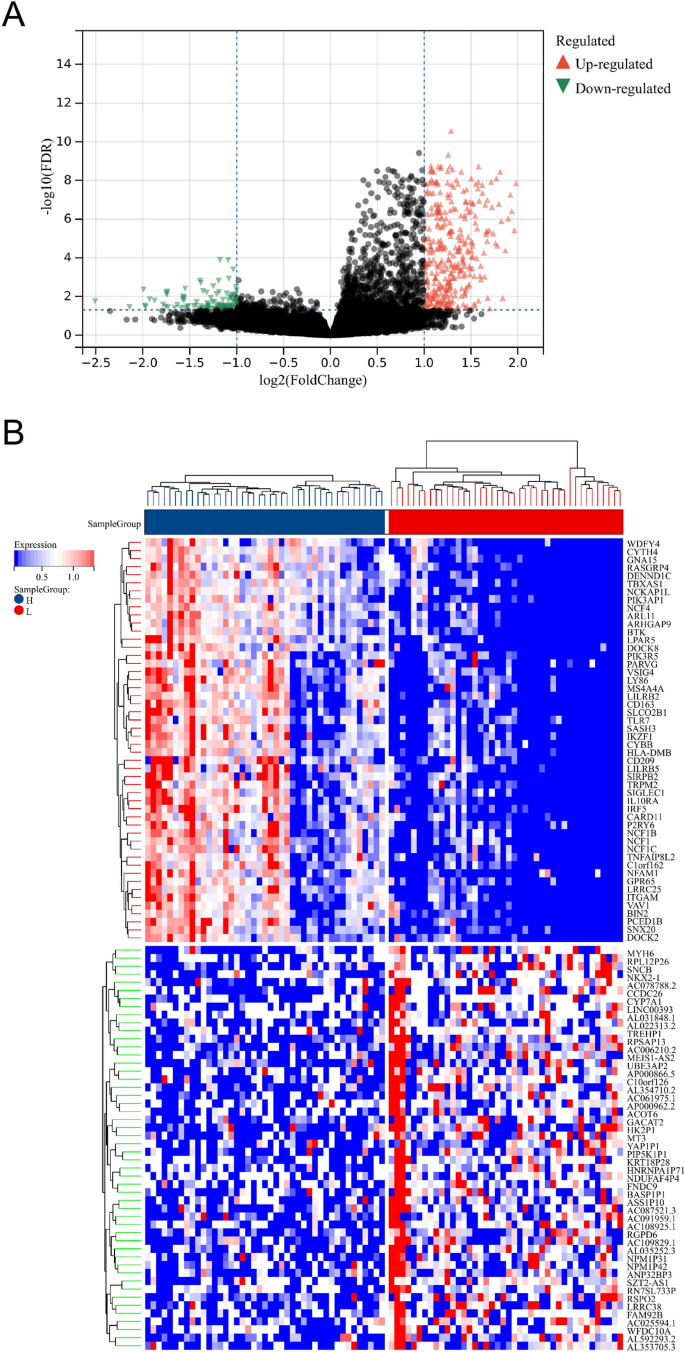
Identificación de DEG entre los subgrupos LML y HML. (A) El gráfico del volcán representó los DEG entre los subgrupos LML y HML. Los puntos verdes representaban genes regulados negativamente, mientras que los puntos rojos representaban genes regulados positivamente. (B) El gráfico del mapa de calor representó los 50 DEG principales entre los subgrupos LML y HML.
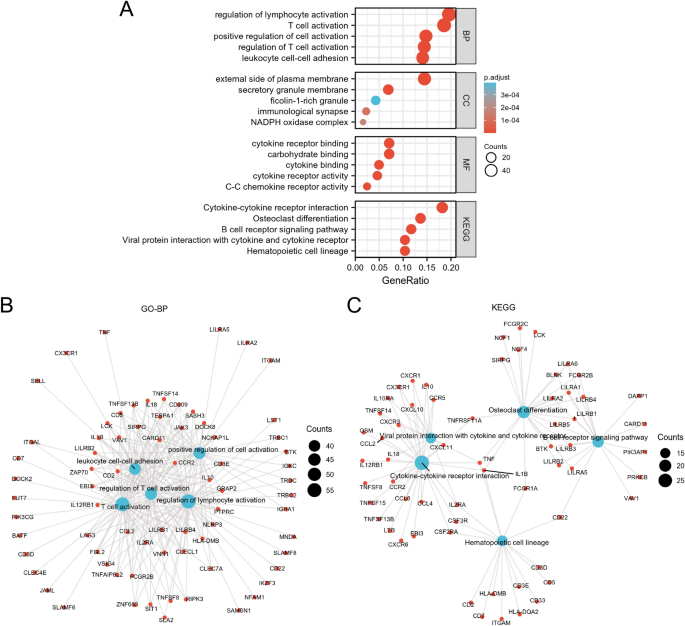
Análisis de enriquecimiento de DEG. (A) Los gráficos de burbujas representaron los resultados de GO y KEGG. El diagrama de red representaba términos GO-BP relacionados con el sistema inmunológico y la inflamación (B) y vías KEGG (W.). Los nodos azules representaban términos GO-BP o vías KEGG, los nodos rojos representaban genes implicados en las vías.
Infiltración de inmunocitos
Analizamos el estado inmunológico entre los subgrupos LML y HML para descifrar el microambiente inmunológico en el sistema operativo. Como se muestra en la Figura 4A, en comparación con el grupo HML, la puntuación estromal, la puntuación inmunológica y la puntuación ESTIMATE disminuyeron en el grupo LML (PAG<0,01), mientras que la pureza del tumor aumentó significativamente en el grupo LML (PAG<0,01). En comparación con el grupo HML, los niveles de células endoteliales, células dendríticas mieloides, linaje monocítico, linaje B, células T y células T CD8 disminuyeron significativamente en el grupo LML (PAG< 0,05) (Figura 4B).
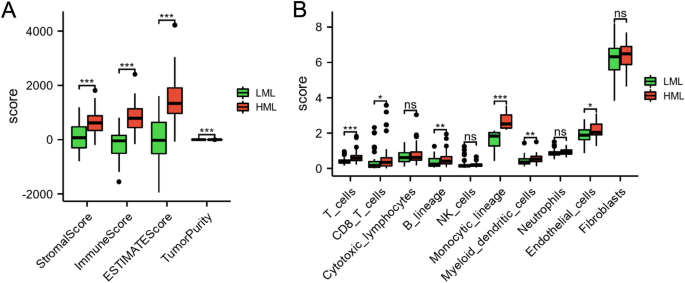
El panorama de los niveles de infiltración de inmunocitos en los dos subgrupos. (A) Comparaciones de puntuación estromal, puntuación inmunológica, puntuación ESTIMATE y pureza tumoral entre los subgrupos LML y HML. (B) Comparaciones de los niveles de infiltración de inmunocitos entre los subgrupos LML y HML. *PAG<0,05, **PAG< 0,01 y ***PAG< 0,001.
Construcción y evaluación del modelo pronóstico de riesgo.
Después del análisis de regresión de Cox univariado, seleccionamos 122 genes relacionados con la supervivencia general (Tabla S1). Posteriormente, el análisis de regresión de LASSO Cox identificó ocho genes (CCDC26, TERT, GJA5, KRT18P28, LILRA6, PDE1B, CD180 e IL2RA) para el análisis de regresión de Cox multivariado (Fig. 5A, B). Finalmente, se seleccionaron y utilizaron tres genes importantes relacionados con la supervivencia general (TERT, IL2RA y CCDC26) para establecer el modelo de pronóstico (Fig. 5C).
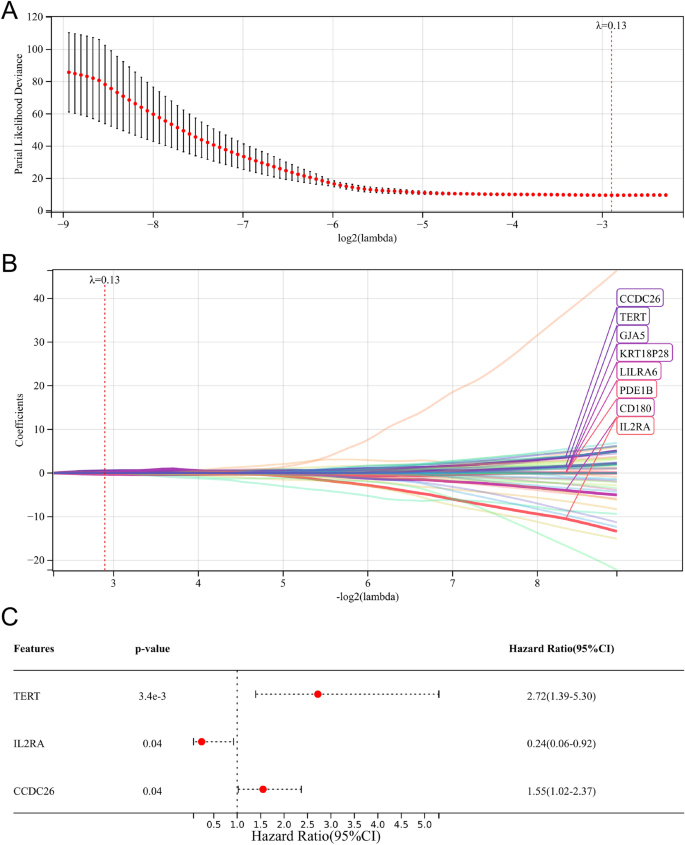
Establecimiento de un modelo pronóstico asociado a ML. (A, B) Análisis de regresión LASSO. (W.) Análisis de regresión multivariada de Cox.
En el conjunto de datos TARGET, el nivel de expresión de TERT y CCDC26 se reguló negativamente en el grupo de bajo riesgo, mientras que la expresión de IL2RA se reguló positivamente en el grupo de bajo riesgo. Además, el grupo de alto riesgo tenía una menor proporción de personas vivas (Fig. 6A). Los pacientes con SG en el grupo de bajo riesgo mostraron una mayor supervivencia general que aquellos en el grupo de alto riesgo (Fig. 6B, PAG< 0,001). El AUC para este modelo de pronóstico fue de 0,8 al año, 0,87 a los 3 años y 0,85 a los 5 años (Fig. 6C), este resultado indicó que el modelo de pronóstico tuvo un buen rendimiento diagnóstico para pacientes con SG. También utilizamos el conjunto de datos GSE21257 para verificar las características de diagnóstico y pronóstico del modelo de riesgo. 7A, el nivel de expresión de TERT y CCDC26 se reguló negativamente en el grupo de bajo riesgo, mientras que la expresión de IL2RA se reguló positivamente en el grupo de bajo riesgo. Además, el grupo de alto riesgo tenía una menor proporción de personas vivas. Los pacientes con SG en el grupo de bajo riesgo mostraron una mayor supervivencia general que aquellos en el grupo de alto riesgo (PAG= 0,025) (Figura 7B). El AUC para este modelo de pronóstico fue de 0,84 al año, 0,67 a los 3 años y 0,68 a los 5 años (Fig. 7C). Estos resultados fueron consistentes con los resultados del conjunto de datos TARGET.
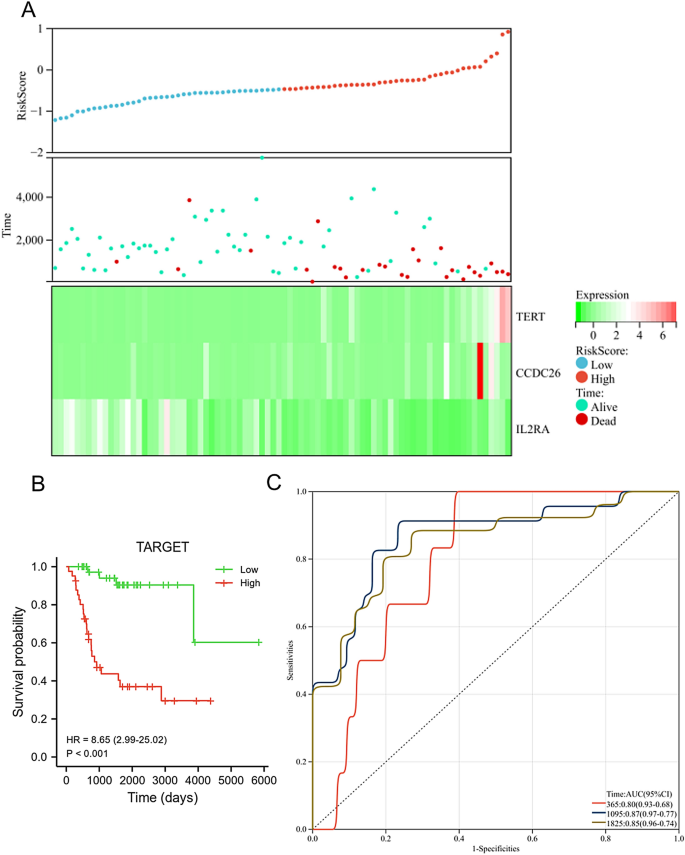
Evaluación del modelo de pronóstico en la base de datos TARGET. (A) El nivel de expresión de TERT, CCDC26 e IL2RA (abajo), el estado de supervivencia (medio) y la distribución de las puntuaciones de riesgo entre los grupos de riesgo bajo y alto (arriba). (B) El análisis de supervivencia mostró la diferencia entre los grupos de bajo y alto riesgo. (W.) Análisis de curvas ROC dependientes del tiempo del modelo de pronóstico.
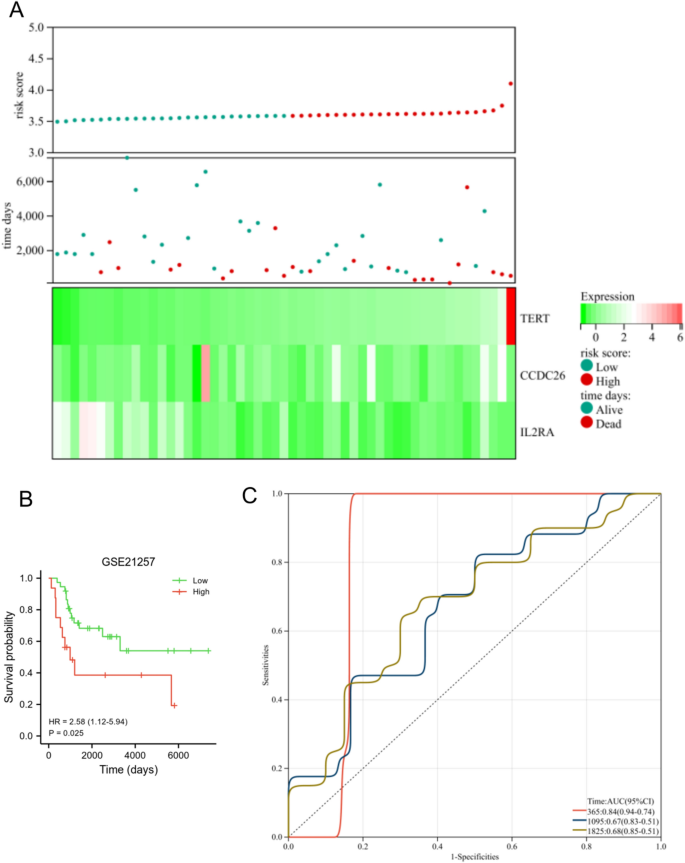
Validación del modelo de pronóstico en el conjunto de datos GSE21257. (A) El nivel de expresión de TERT, CCDC26 e IL2RA (abajo), el estado de supervivencia (medio) y la distribución de las puntuaciones de riesgo entre los grupos de riesgo bajo y alto (arriba). (B) El análisis de supervivencia mostró la diferencia entre los grupos de bajo y alto riesgo. (W.) Análisis de curvas ROC dependientes del tiempo del modelo de pronóstico.
Además, se estableció un nomograma para ayudar aún más a predecir el pronóstico de los pacientes con OS (Fig. 8A). Los resultados de la predicción del nomograma fueron muy consistentes con la observación de pacientes con OS según la curva de calibración del nomograma (Fig. 8B).
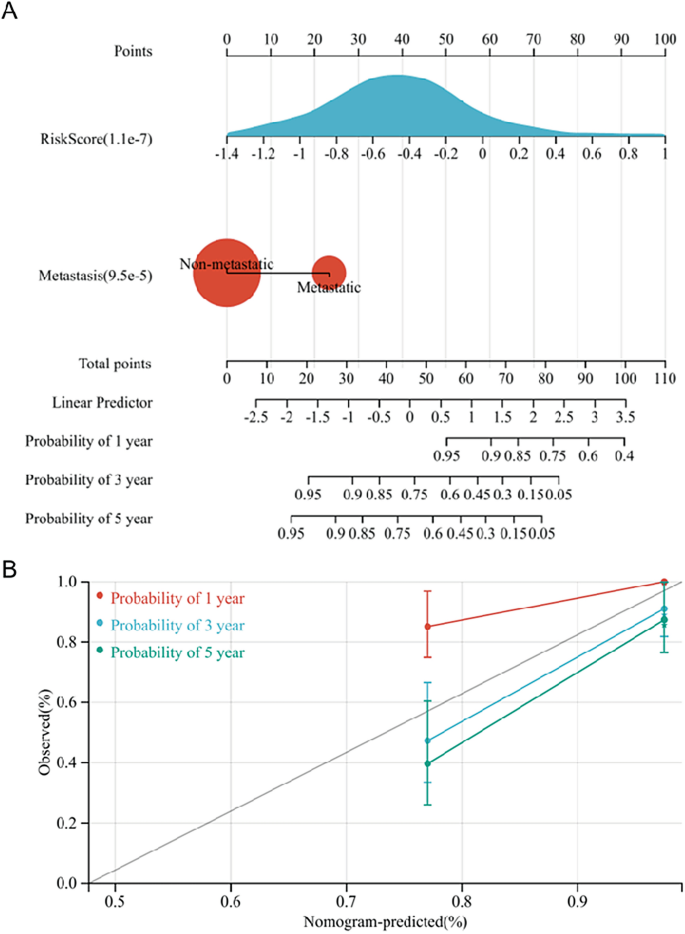
Establecimiento del nomograma. (A) Se utilizaron metástasis y puntuación de riesgo para establecer el nomograma. (B) Curva de calibración del nomograma.
Análisis de interacción de genes pronósticos.
Al emplear la base de datos GeneMANIA, construimos con éxito una red de interacción de proteínas para los genes característicos (TERT e IL2RA). A través de este análisis, descubrimos un total de 20 genes que interactúan con los genes característicos (Fig. 9A). Se realizó un análisis de enriquecimiento funcional en estos 22 genes. Los resultados obtenidos del análisis de enriquecimiento demostraron que estos genes están predominantemente relacionados con la organización de los telómeros, el mantenimiento de los telómeros, la infección por el virus de la leucemia de células T humana 1, la diferenciación de las células Th1 y Th2, etc.
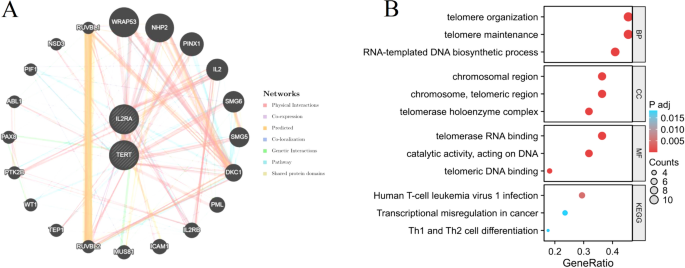
Análisis de interacción de genes característicos. (A) La red de coexpresión de genes característicos. (B) Análisis de enriquecimiento funcional de genes coexpresados mediante GO y KEGG.
Asociación entre genes relacionados con el modelo de riesgo y microambiente tumoral (TME)
Realizamos un análisis de los niveles de expresión de los genes TERT e IL2RA en células asociadas con el microambiente tumoral dentro del sistema operativo utilizando la base de datos TISCH. Nuestros hallazgos demostraron que IL2RA mostró un mayor nivel de infiltración en cDC1, monocitos y células M2 (Fig. 10).
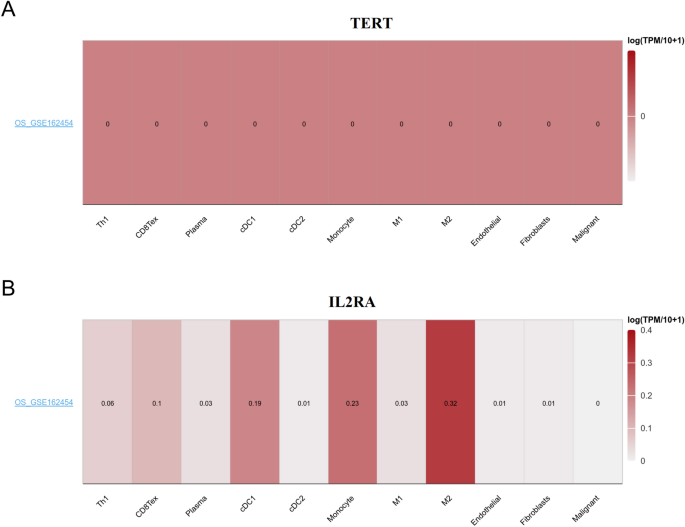
Los genes asociados con modelos de riesgo se expresaron en células relevantes para el microambiente del tumor. Niveles de expresión TERT (A) y IL2RA (B) en celdas relacionadas con el microambiente del sistema operativo se visualizaron utilizando un mapa de calor en el conjunto de datos GSE162454.
El modelo de pronóstico tiene la capacidad de distinguir pacientes con OS metastásico
Como se muestra en la Figura 11A, en comparación con el grupo de alto riesgo (TARGET, 58,54 % y GSE21257, 17,25 %), se observaron más casos sin metástasis (TARGET, 90,25 % y GSE21257, 58,34 %) en el grupo de bajo riesgo. Además, en comparación con el grupo metastásico, la puntuación de riesgo fue menor en el grupo no metastásico (PAG<0,01, Figura 11B,C). Además, los resultados del análisis ROC mostraron que el rendimiento diagnóstico del modelo de pronóstico para la predicción de metástasis fue de 0,659 y 0,705 en TARGET y GSE21257, respectivamente (Fig. 11D, E). Estos hallazgos mostraron que el modelo de riesgo podría predecir la metástasis en pacientes con OS.
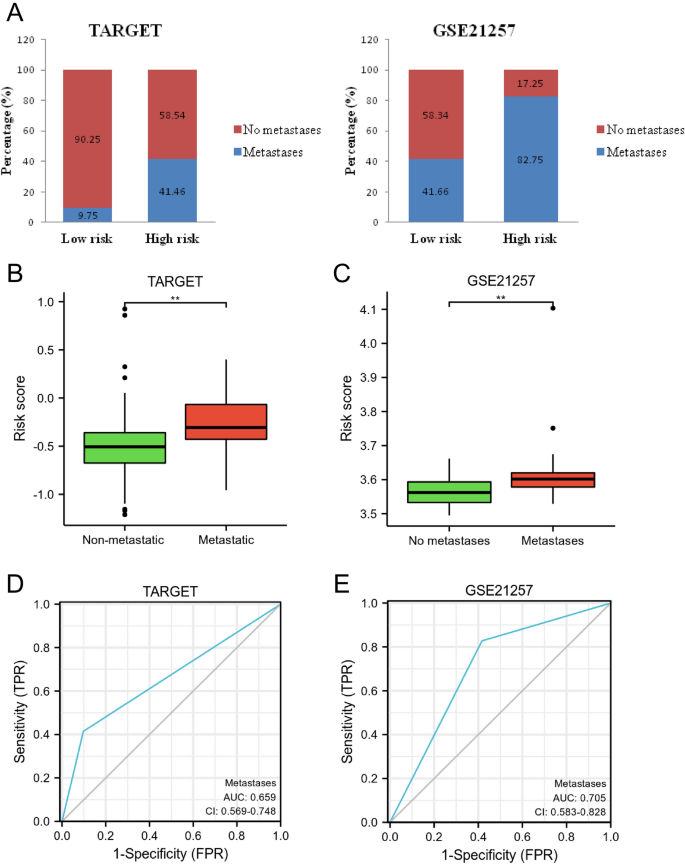
Evaluación de la capacidad del modelo pronóstico para predecir metástasis en pacientes con OS. (A) Comparaciones de casos con metástasis y sin metástasis en los grupos de bajo y alto riesgo. (B) Comparaciones de puntuaciones de riesgo en grupos metastásicos y no metastásicos. (W.) Análisis ROC para el rendimiento diagnóstico en la predicción de metástasis del sistema operativo.




